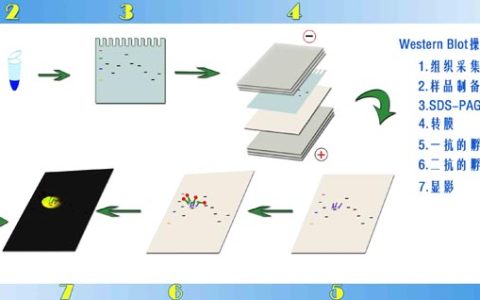
RNA免疫共沉淀方法:RIP is an antibody-based technique used to map in vivo RNA-protein interactions and RNA modifications such as m6A and ac4C. The RNA binding protein (RBP) of interest is immunoprecipitated together with its associated RNA to identify bound transcripts (mRNAs, non-coding RNAs, or viral RNAs). Transcripts are detected by real-time PCR, microarrays, or sequencing.
The fields of epigenetics and RNA biology have recently seen a huge increase in the interest in different RNA roles and functions. Beyond transcription and subsequent translation, it has been observed that there is much more to the function of RNA. For example, RNA-protein interactions can modulate mRNA and noncoding RNA function. This new appreciation for the potential of RNA has lead to the development of novel methods allowing researchers to map RNA-protein interactions. RIP is one such protocol allowing the study of the physical association between individual proteins and RNA molecules.
The following RIP protocol is adapted from Khalila et al. (2009), Hendrickson et al. (2009), Hendrickson et al. (2008), and Rinn et al. (2007).
Protocol summary
1. Harvest cells (optional treatment of cells with formaldehyde to cross-link in vivo
protein-RNA complexes)
2. Isolate nuclei and lyse nuclear pellets
3. Shear chromatin
4. Immunoprecipitate the RNA binding protein (RBP) of interest together with the bound RNA
5. Wash off unbound material
6. Purify RNA that is bound to immunoprecipitated RBP
7. Reverse transcribe RNA to cDNA and analyze by qPCR, microarray, or sequencing
RIP protocol
Download a printable version of this protocol
1. Cell harvesting
1.1. Grow cells to confluency and treat as required for the experiment.
1.2. If a cross-linking step is required, this will require optimization of the fixation time.
View the cross-linking section of our ChIP protocol.
1.3. Harvest cells by trypsinization and resuspend in PBS (e.g., 107 cells in 2 mL PBS), freshly prepared nuclear isolation buffer (2 mL), and water (6 mL). Keep on ice for 20 min (with frequent mixing).
One or more negative controls should be maintained throughout the experiment, e.g., no antibody sample or immunoprecipitation from knockout cells or tissue. Knockdown cells are not recommended for negative control experiments.
2. Nuclei isolation and lysis pellets
2.1. Pellet nuclei by centrifugation at 2,500 g for 15 min.
2.2. Resuspend nuclear pellet in freshly prepared RIP buffer (1 mL).
Avoid contamination using RNase-free reagents such as RNase-free tips, tubes, and reagent bottles; also use ultrapure distilled, DNase-free, RNase-free water to prepare buffers and solutions.
3. Chromatin shearing
3.1. Split resuspended nuclei into two fractions of 500 µL each (for mock and IP).
3.2. Mechanically shear chromatin using a dounce homogenizer with 15–20 strokes.
Different cell lines might require optimization of shearing conditions.
3.3. Pellet nuclear membrane and debris by centrifugation at 13,000 rpm for 10 min.
Freeze an aliquot of lysate in liquid nitrogen for reference RNA isolation.
4. RNA immunoprecipitation
4.1. Add antibody to the protein of interest (2–10 ug) to the supernatant (6–10 mg) and incubate for 2 h (to overnight) at 4oC with gentle rotation.
4.2. Add protein A/G beads (40 µL) and incubate for 1 h at 4oC with gentle rotation.
The amount of antibody added and the incubation time might need to be optimized depending on the protein of interest and antibody. If an antibody is working in IP, this is a good indication that it will work in RIP.
5. Washing off unbound material
5.1. Pellet beads at 2,500 rpm for 30 s, remove supernatant and resuspend beads in 500 µL RIP buffer
Stringent washing of protein A/G bead pellets is important and might need to be optimized.
5.2. Repeat for a total of three RIP washes, followed by one wash in PBS
Freeze 5% of the beads for SDS-PAGE analysis after the second wash (e.g., use 5 µL of bead slurry if you have 100 µL total bead slurry volume).
6. Purification of RNA that was bound to immunoprecipitated RBP
1. Isolate coprecipitated RNAs by resuspending beads in TRIzol RNA extraction reagent (1 mL) according to manufacturer’s instructions. Further information can be found in our RNA isolation protocol.
2. Elute RNA with nuclease-free water (e.g., 20 µL).
Add approximately 15–25 µL (depending on yield) of either DEPC treated TE buffer or water to the RNA pellet. Eluted RNA can be stored at -80°C.
3. Protein isolated by the beads can be detected by western blot analysis. Further information can be found in our western blot protocol.
If a cross-linking step has been used (1.2), the cross-link should now be reversed. View the reverse cross-links section of our ChIP protocol for details.
7. Reverse transcription (RT) of RNA to cDNA and analysis
1. Reverse transcribe DNAse treated RNA according to manufacturer instructions.
2. Analyze by qPCR of cDNA if the target is known. If the target is not known, the creation of cDNA libraries, microarrays, and sequencing can be used for analysis.
The control experiments should give no detectable products after PCR amplification, and high-throughput sequencing of these control libraries should return very few unique sequences.
Reagents
- 1.28 M sucrose
- 40 mM Tris-HCl pH 7.5
- 20 mM MgCl2
- 4% Triton X-100
- 150 mM KCl
- 25 mM Tris pH 7.4
- 5 mM EDTA
- 0.5 mM DTT
- 0.5% NP40
- 100 U/ml RNAase inhibitor SUPERase•in™ (add fresh each time)
- Protease inhibitors (add fresh each time)
References
Khalila AM, Guttman M, Huarte M, Garbera M, Rajd A, Morales DR, Thomas K, Pressera A, Bernstein BE, Oudenaarden AV, Regeva A, Lander ES, and Rinn JL (2009). Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. PNAS 106, 11667–72.
Hendrickson DG, Hogan DJ, McCullough HL, Myers JW, Herschlag D, Ferrell JE, and Brown PO (2009). Concordant Regulation of Translation and mRNA Abundance for Hundreds of Targets of a Human microRNA. PLoS Biology 7 (11), 2643.
Hendrickson DG, Hogan DJ, Herschlag D, Ferrell JE, and Brown PO (2008). Systematic Identification of mRNAs Recruited to Argonaute 2 by Specific microRNAs and Corresponding Changes in Transcript Abundance. PLoS One 3 (5), 2126.
Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, and Chang HY (2007). Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129, 1311–1323.
本站原创,如若转载,请注明出处:https://www.ouq.net/2253.html
 微信打赏,为服务器增加50M流量
微信打赏,为服务器增加50M流量  支付宝打赏,为服务器增加50M流量
支付宝打赏,为服务器增加50M流量