
基于同源性的搜索通常用于从宏基因组中发现移动遗传元件 (MGE),但它在很大程度上依赖于数据库中的参考基因组。 该方法可以在不使用参考数据库的情况下从组装的人宏基因组序列中提取 CRISPR 靶向序列。 我们描述了宏基因组重叠群的组装、CRISPR 直接重复序列和间隔区的提取、原型间隔区的发现以及使用基于图的方法提取富含原型间隔区的区域,可以提取许多特征/非特征的 MGE。
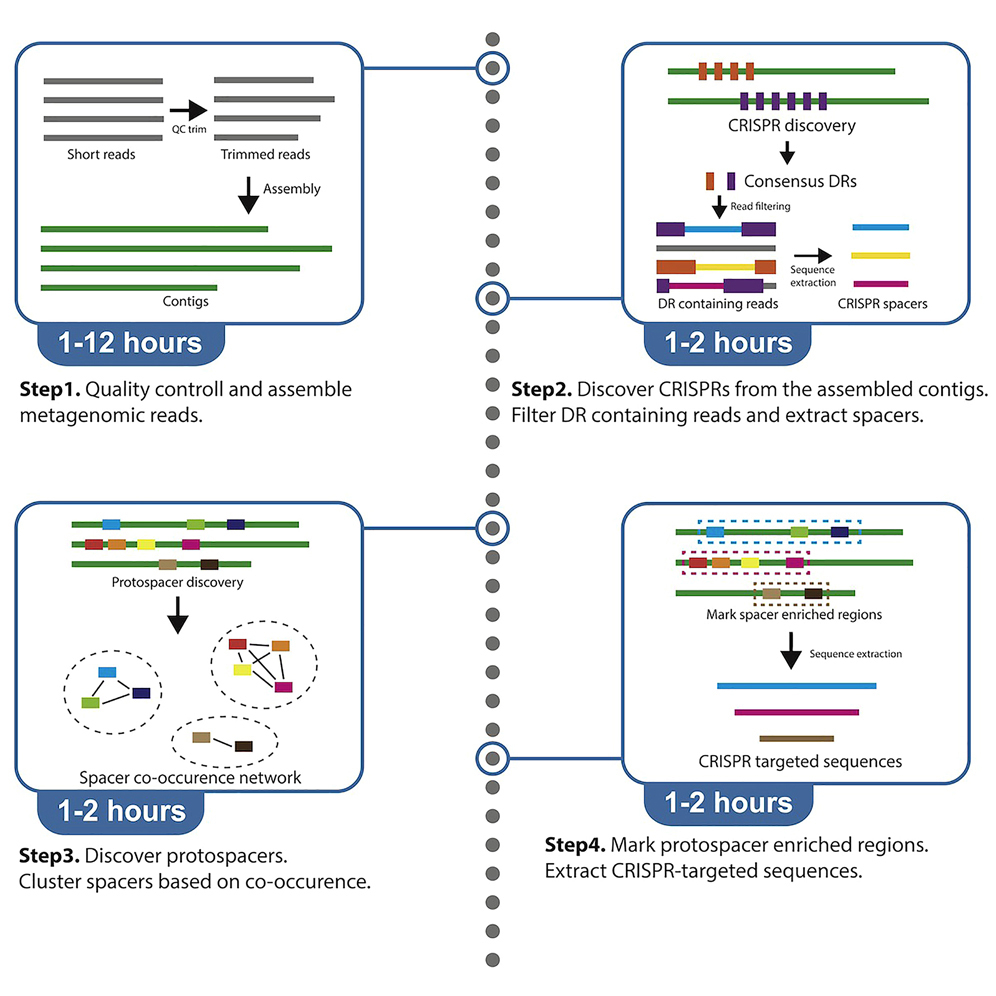
Preprocess raw paired FASTQ files
This process is quality control of the input sequences.
- Trim poor-quality bases, adapters, and PhiX spikes.
$ bbduk.sh -Xmx100g t=8 in1=r1.fastq.gz in2=r2.fastq.gz out=trimmed.fastq.gz ftm=5 qtrim=rl trimq=20 ftl=15 ref=adapters,phix ktrim=r k=23 mink=11 hdist=1 tpe tbo maq=20
- Remove human-derived reads.
$ bbmap.sh -Xmx100g t=8 ref={path to the human reference FASTA file} in=trimmed.fastq.gz outu=decontaminated.fastq.gz interleaved=t minratio=0.9 maxindel=3 bwr=0.16 bw=12 fast=t minhits=2 qtrim=r trimq=10 untrim=t idtag=t printunmappedcount=t kfilter=25 maxsites=1 k=14
- Correct errors in the reads.
$ tadpole.sh threads=8 -Xmx100g interleaved=t in=decontaminated.fastq.gz out=ecc.fastq.gz mode=correct
Metagenome assembly
Assemble metagenome contigs from the preprocessed FASTQ files.
- Assemble preprocessed FASTQ file using SPAdes.
$ spades.py -t 8 -m 100 --meta --only-assembler --12 ecc.fastq.gz -o {path to spades output directory}
- Assign unique identifiers to all assembled contigs.
$ cd {path to spades output directory}
$ id=$(openssl rand -hex 12)
awk -v id=${id} '/ˆ>/ {n++; print ">contig_"id"_"n;} !/ˆ>/{print}' scaffolds.fasta > scaffolds.renamed.fasta
Extraction of CRISPR direct repeats from the assembled contigs
From the assembled contigs, discover CRISPR consensus direct repeats.
- Run CRISPRDetect to discover consensus CRISPR direct repeats.
$ CRISPRDetect.pl -q 0 -f scaffolds.renamed.fasta -o crispr_detect_output -array_quality_score_cutoff 2
- From the output gff file, extract direct repeats, and convert them into a FASTA file.
$ grep 'repeat_region' crispr_detect_output.gff | cut -f 9 | tr ';' '∖n' | egrep 'ˆNote=' | cut -f 2 -d '=' | awk '{n++; printf(">DR_%i∖n%s∖n", n, $0)}' > crispr_dr.fasta
- After discovering all direct repeats from each assembled FASTA file, we merge them into a single file, assign unique identifiers, and remove redundancy.
$ cat {all crispr dr fasta files} > merged_crispr_dr.fasta
$ awk '/ˆ>/ {n++; print ">dr_"n; } !/ˆ>/ {print}' merged_crispr_dr.fasta > merged_crispr_dr.renamed.fasta #giving new unique identifiers
$ cd-hit-est -S 1 -c 1 -i merged_crispr_dr.renamed.fasta -o merged_crispr_dr.repr.fasta
Extraction of CRISPR spacers from the preprocessed reads
Here, we return to the preprocessed FASTQ files. From them, we extract CRISPR spacers from reads containing CRISPR direct repeats.
- Extract direct repeat-containing reads. This initial filtering step significantly reduces the number of reads, effectively speeding up the following spacer extraction processes.
$ bbduk.sh in=ecc.fastq.gz ref=merged_crispr_dr.repr.fasta outm=dr_containing_reads.fastq.gz interleaved=t k=21 hdist=1 rename=t
- Second filter to mask the direct repeats in the reads.
$ bbduk.sh in=dr_containing_reads.fastq.gz ref=merged_crispr_dr.repr.fasta kmask=R k=19 hdist=1 out=dr_masked.fastq mink=15
- Extract spacers from the masked reads using our python script.
$ {path to the virome_scripts directory}/pipeline_scripts/extract_spacers.py -s {sample name} dr_masked.fastq > spacers.fasta
- Merge all discovered spacers, assign unique identifiers, and remove redundancy.
$ cat {all spacer fasta files} > all_spacers.fasta
$ awk '/ˆ>/ {sub(/ˆ[ˆ[:space:]]+[[:space:]]/, ""); n++; printf(“>spacer_"n"∖t"$0);} !/ˆ>/ {print}' all_spacers.fasta > all_spacers.renamed.fasta #giving new unique identifiers
$ cd-hit-est -T 8 -M 10000 -d 0 -sf 1 -s 0.9 -c 0.98 -i all_spacers.renamed.fasta -o all_spacers.repr.fasta
Protospacer discovery
Here, we discover protospacers by aligning the spacer sequences to CRISPR-masked contigs.
- Search CRISPR direct repeats from the contigs.
$ makeblastdb -dbtype nucl -in scaffolds.renamed.fasta
$ blastn -evalue 1e-5 -task ‘blastn-short’ -outfmt 6 -num_threads 8 -query merged_crispr_dr.repr.fasta -db scaffolds.renamed.fasta > dr.blastn
- Mask sequences around direct repeat aligned regions.
$ samtools faidx scaffolds.renamed.fasta
$ awk ‘BEGIN{FS="∖t"; OFS="∖t"} $10<$9{t=$9; $9=$10; $10=t} {print $2,$9–1,$10}' dr.blastn | sort -k1,1 -k2,2n | bedtools merge -i /dev/stdin -d 60 | bedtools slop -b 60 -i /dev/stdin -g scaffolds.renamed.fasta.fai | bedtools maskfasta -bed /dev/stdin -fi scaffolds.renamed.fasta -fo crispr_masked.fasta
- Align spacers to the masked contigs.
$ makeblastdb -dbtype nucl -in crispr_masked.fasta
$ blastn -evalue 1e-5 -perc_identity 93 -task ‘blastn-short’ -outfmt 6 -num_threads 8 -query all_spacers.repr.fasta -db crispr_masked.fasta > spacers.blastn
- Merge all discovered protospacers into a single BED-formatted file.
$ cat {all spacer blastn output files} | awk ‘BEGIN{FS="∖t"; OFS="∖t"} $10<$9{t=$9; $9=$10; $10=t} {print $2,$9–1,$10,$1}' | sort -k1,1 -k2,2n > all_protospacers.bed
Spacer clustering based on co-occurrence
Here, we cluster spacers using a co-occurrence network calculated from the spacer alignment result. From this network, we discover the graph communities, representing subsets of spacers that corresponding protospacers co-occur together across the assembled contigs. We extract the protospacer-enriched regions using these graph communities, i.e., spacer clusters. This process effectively reduces the false-positive ratio by excluding lone or randomly scattered protospacers.
- Initial clustering is based on distance.
$ bedtools cluster -d 50000 -i all_protospacers.bed | awk ‘BEGIN{FS="∖t"; OFS="∖t"} {print $1"_cluster_"$5,$2,$3,$4}' > initial_cluster.bed
- Calculate a weighted graph from the spacer co-occurrence and generate an abc formatted file.
$ {path to the virome_scripts directory}/graph_clustering/generate_abc_edgefile.py initial_cluster.bed > protospacers.abc
- Convert the abc file into an mci format.
$ mcxload -abc protospacers.abc --stream-mirror -write-tab data.tab -o protospacers.mci
- Run an mcl program to discover graph communities.
$ mcl protospacers.mci -I 4 -pi 0.4 -te 8
- Convert the mcl output to a tabular format.
$ mcxdump -icl out.protospacers.mci.I40pi04 -tabr data.tab > protospacers_cluster.tab
- Mark CRISPR-targeted regions using the clustering result and the merged protospacer bed file.
$ {path to the virome_scripts directory}/graph_clustering/mark_crispr_targeted.py all_protospacers.bed protospacers_cluster.tab > crispr_targeted.bed
- Merge the regions to rescue the fragmented clusters.
$ sort -k1,1 -k2,2n crispr_targeted.bed > crispr_targeted.sorted.bed
$ bedtools merge -d 1000 -i crispr_targeted.sorted.bed -o collapse -c 4 > crispr_targeted.merged.bed
- Extract CRISPR-targeted regions from the assembled contigs.
$ samtools faidx scaffolds.renamed.fasta
$ cut -f 1,2 scaffolds.renamed.fasta.fai | sort -k 1,1 | join -t $'∖t' - <(sort -k 1,1 crispr_targeted.merged.bed) | awk 'BEGIN{OFS="∖t"; FS="∖t"} $4>$2{$4=$2} {print $1,$3,$4,$5}' | bedtools getfasta -fi scaffolds.renamed.fasta -bed /dev/stdin -fo crispr_targeted.fasta
本站原创,如若转载,请注明出处:https://www.ouq.net/2124.html
 微信打赏,为服务器增加50M流量
微信打赏,为服务器增加50M流量  支付宝打赏,为服务器增加50M流量
支付宝打赏,为服务器增加50M流量 
